How to Know if Fastq Reads Are Trimmed
BCL to FASTQ conversion
Introduction
Illumina sequencing technology uses cluster generation and sequencing by synthesis chemical science to sequence millions or billions of clusters on a menses cell, depending on the sequencing platform. During sequencing, for each cluster, base calls are fabricated and stored for every cycle of sequencing by the Real-Time Analysis (RTA) software on the instrument. RTA stores the base call data in the class of private base call (or BCL) files. When sequencing completes, the base calls in the BCL files must be converted into sequence information. This procedure is called BCL to FASTQ conversion.
Multiplex sequencing
Multiplex sequencing allows large numbers of libraries to be pooled and sequenced simultaneously during a single run on a high-throughput instrument. With multiplexed libraries, unique index sequences are added to each Dna fragment during library preparation so that each read can be identified and sorted before final information assay. Pooling samples exponentially increases the number of samples analysed in a single run, without drastically increasing cost or time.
Gains in throughput from multiplexing come with an added layer of complication, every bit sequencing reads from pooled libraries demand to be identified and sorted computationally in a process called demultiplexing before concluding data assay. The phenomenon of index misassignment between multiplexed libraries is a known issue that has impacted NGS technologies from the time sample multiplexing was adult.
BCL and FASTQ file formats
Binary Base Call (BCL) files are the raw data files generated past the Illumina sequencers. The Real Time Analysis (RTA) software writes the base of operations and the conviction in the call equally a quality score to base call (.bcl) files. Equally the name implies this is done in real time, i.e. for every cycle of the sequencing run a phone call for every location identified on the flow jail cell (tiles and lanes) is added. BCL files are stored in binary format and correspond the raw information output of a sequencing run.
The FASTQ is a text-based sequence file format that is generated from the BCL file that stores both raw sequence data and quality scores. For a unmarried-read run, one FASTQ file is created for each sample per flow cell lane. For a paired-cease run, 2 FASTQ files are created for each sample for each lane.

Each entry in a FASTQ files consists of 4 lines:
- A sequence identifier with information well-nigh the sequencing run and the cluster. The exact contents of this line vary based on the BCL to FASTQ conversion software used.
- The sequence (the base calls; A, C, T, K and North).
- A separator, which is simply a plus (+) sign.
- The base phone call quality scores. These are Phred +33 encoded, using ASCII characters to represent the numerical quality scores.
Conversion tools
CASAVA
Illumina's CASAVA software bundle was used for bcl-to-fastq conversion (among other things that these package provides). With the newer sequencing instruments came compressed bcl files that CASAVA could not handle. Illumina now provides a bcl2fastq software package which only includes the components of CASAVA required for FASTQ conversion and which can besides deal with compressed bcl files.
bcl2fastq
The Illumina bcl2fastq Conversion Software demultiplexes sequencing data and converts BCL files into FASTQ files. bcl2fastq v1.8.4 is derived from CASAVA and it's fully compatible with the alignment and variant calling components of CASAVA 1.8. The bcl2fastq v1.eight.4 tin can exist used for converting from any Illumina sequencing organization running RTA v1.18.54, or earlier. For newer versions of RTA, bcl2fastq2 v2.20 has to be used.
bcl2fastq v1.8.four
Bcl conversion and demultiplexing is washed in a single pace, and generates a new directory in the Run folder, which contains all of the demultiplexed compressed FASTQ files.
Folder and File Naming
The top level run binder proper name is generated using three fields to identify the <ExperimentName>, separated by underscores. For example: YYMMDD_<machine_name>_NNNN. Yous should not deviate from the run folder naming convention, equally this may cause the software to stop.
- The kickoff field is a half-dozen-digit number specifying the date of the run.
- The second field specifies the name of the sequencing machine. It may consist of any combination of upper or lower case messages, digits, or hyphens, but may non comprise any other characters (especially not an underscore).
- The third field is a four-digit counter specifying the experiment ID on that instrument.
Bcl Conversion Input Files
Demultiplexing needs a BaseCalls directory and a sample sheet to outset a run.
BaseCalls Directory
Demultiplexing requires a BaseCalls directory containing the binary base phone call files (BCL files) every bit generated past RTA, OLB (Off-Line Basecaller), or RTAOLB. The BCL to FASTQ converter needs the post-obit input files from the BaseCalls directory:
- BCL files.
- *.stats files.
- *.filter files.
- *.control files
- *.clocs, *.locs, or *_pos.txt files. The BCL to FASTQ converter determines which blazon of position file it looks for based on the RTA version that was used to generate them.
- RunInfo.xml file. The RunInfo.xml is at the top level of the run folder.
- config.xml file. RTA is configured to re-create these files off the instrument computer car to the BaseCalls directory on the assay server.
Sample sheet
The sample sheet (SampleSheet.csv file) directs the software how to assign reads to samples, and samples to projects. The sample canvass specifies for every index in every lane which sample and which project it belongs to. Lanes with samples that were not indexed can also be assigned to samples and projects using the sample sheet. It is needed only if the demultiplexing needs to be preformed. The sample sheet should be located in the BaseCalls directory of the run binder. Alternatively the location of the file can be specified with the — sample-sheet pick.
You can create, open, and edit the sample sheet in Excel, but it is easier and recommended to use the Illumina Experiment Director (IEM) to create them. IEM is a sorcerer-driven application that guides yous through the creation and setup of your sample sheet. More than information can be establish here.

Conversion
Bcl conversion and demultiplexing is configured by script configureBclToFastq.pl.
The standard way to run bcl conversion and demultiplexing is to get-go create the necessary Makefiles, which configure the run. Then you run make on the generated files, which executes the calculations.
One example of creating make file - run the control:
configureBclToFastq.pl /
— input-dir <path_to_BaseCalls_dir> /
— output-dir <path_to_output_dir> /
— sample-sheet <path_to>/SampleSheet.csv
This volition create the named output directory containing a Makefile which performs the actual conversion; to run, 'cd' to the output directory and and then run make.
In that location is a big number of options that can be used with this command. Information on all of theme can be constitute in the software guide.
Some of them are:
- — i, — — input-dir <Path to a BaseCalls directory>. Defaults to current directory.
- — o, — — output-dir <Path to demultiplexed output>.Defaults to <run_folder>/Unaligned.
- — s, — — sample-sheet <Path to sample sheet file>. Defaults to <input_dir>/SampleSheet.csv. Needed just for demultiplexing.
- — mismatches <n, n, n…>. Number of mismatches allowed for each read; the default is nada (recommended for samples without multiplexing), 1 mismatch is recommended for multiplexed samples with tags of length 6 bases.
- — ignore-missing-bcl. Interpret missing *.bcl files as no call.
Bcl Conversion Output Folder
The Bcl Conversion output directory has the following characteristics:
- The project and sample directory names are derived from the sample sheet.
- The Unaligned/Basecall_Stats_FCID/Demultiplex_Stats.htm file shows where the sample information are saved in the directory structure.
- The Undetermined_indices directory contains the reads with an unresolved or erroneous index.
- If no sample sheet exists, the software generates a project directory named after the flow cell, and sample directories for each lane.
Output FASTQ Files
bcl2fastq converts *.bcl files into FASTQ files, which can exist used as sequence input for alignment. The files are located in the Unaligned/Project_<ProjectName>/Sample_<SampleName> directories. Illumina FASTQ files use the following naming scheme:
<sample name>_<barcode sequence>_L<lane>_R<read number>_<set number>.fastq.gz
Upgrade to bcl2fastq2
Some key features that are only available in bcl2fastq2 v2.17 and later are:
- Demultiplex runs from all sequencing platforms running RTA v1.18.54 and after.
- Demultiplex runs with mixed index types (eg, 6 bp and 8 bp) in dissimilar lanes in the sample sheet in a single analysis.
- Handle unique molecular identifiers (UMIs) in FASTQ files via a sample sheet setting.erences between bcl2fastq v1.eight.four and bcl2fastq2 v2.17 and later
As well, one of the fundamental differences is the required sample sheet format. To exist sure that the format is right always use Illumina Experiment Manager (IEM) for generating sample canvas.
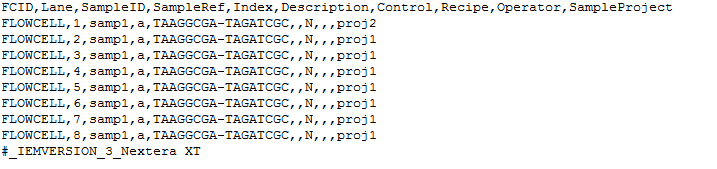

Other differences tin be seen in the table below:

bcl2fastq2 v2.xx
In that location are many similarities with the bcl2fastq v1.8.iv so nosotros will highlight just differences here.
Folder and File Naming
Output folder naming convention depends on the system and can include the aforementioned variables listed in the bcl2fastq v1.8.4. Equally a best practice, give experiments and samples unique names to prevent naming conflicts.
Bcl Conversion Input Files
The following table lists the output files that contain sequencing data. The bcl2fastq2 Conversion Software uses these output files as input.
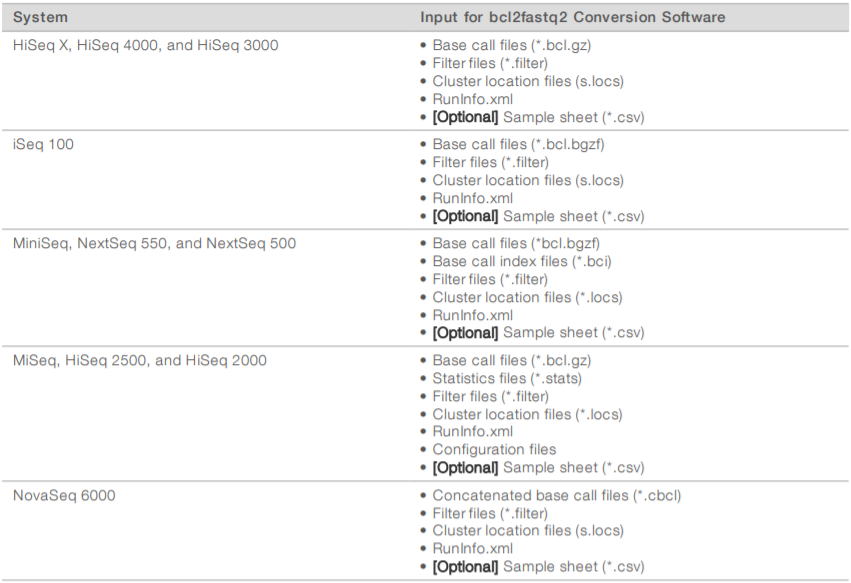
Sample sheet
The new sample sheet format allows specification of more than settings than it was able in the previous version. Information technology has 2 principal sections:
- Settings Section — The software uses the Settings department of the sample sheet to specify adapter trimming, cycle, UMI, and index options.
- Data Section — The software uses columns in the Data department to sort samples and index adapters.
Conversion
The conversion is washed past running a command bcl2fastq.
Add command options to modify the software performance as needed. If you add together options that have a corresponding sample sheet setting, the command-line value overwrites the sample canvas value. Besides options from the previous version (similar specifying the output or input directory), in that location are some new and interesting ones. I of them are options for threads:
- — r, — — loading-threads <n>. Number of threads to load BCL information
- — p, — — processing-threads <n>. Number of threads to procedure demultiplexing data.
- — w, — — writing-threads <n>. Number of threads to write FASTQ information. This number must be lower than number of samples.
For more than options, check the software guide.
Bcl Conversion Output
The bcl2fastq2 Conversion Software v2.20 generates the following files equally output:
- FASTQ files
- InterOp files
- ConversionStats file
- DemultiplexingStats file
- Adapter Trimming file
- FastqSummary and DemuxSummary
- HTML reports
- JavaScript Object Notation (JSON) file
Output FASTQ files
The software writes compressed, demultiplexed FASTQ files to the directory <run folder>\Data\Intensities\BaseCalls.
- If a sample canvas specifies the Sample_Project column for a sample, the software places the FASTQ files for that sample in the directory <run binder>\Data\Intensities\BaseCalls\<Project>. The same project directory contains the files for multiple samples.
- If the Sample_ID and Sample_Name columns are specified only do not match, the FASTQ files reside in a <SampleID> subdirectory where files utilize the Sample_Name value.
- Reads with unidentified index adapters are recorded in 1 file named Undetermined_S0_. If a sample canvass includes multiple samples without specified index adapters, the software displays a missing barcode error and ends the analysis.
FASTQ files are named by the following convention: <samplename>_S<sample number>_L<lane number>_R<read number ane or 2>_001.fastq.gz.
Source: https://medium.com/@marija190396/bcl-to-fastq-conversion-e289852823d0
0 Response to "How to Know if Fastq Reads Are Trimmed"
Post a Comment